Generating reads¶
InSilicoSeq can simulate amplicon reads, or reads from whole metagenome sequencing (the default).
You can specify the type of reads you want to simulate with the --sequence_type option.
InSilicoSeq comes with a set of pre-computed error models to allow the user to easily generate reads from the most popular Illumina instruments:
HiSeq
- MiSeq (optionally, with various quality thresholds):
MiSeq-20
MiSeq-24
MiSeq-28
MiSeq-32
MiSeq-36
NextSeq
NovaSeq
Per example generate 1 million MiSeq reads from a set of input genomes:
curl -O -J -L https://osf.io/thser/download # download the example data
iss generate --genomes SRS121011.fasta --model miseq --output miseq_reads
This will create 2 fastq files, miseq_reads_R1.fastq and miseq_reads_R2.fastq in your current directory, as well as miseq_reads_abundance.txt, a tab-delimited file containing the abundance of each genomes.
InSilicoSeq will use 2 cpus by default. For multithreading, use --cpus:
curl -O -J -L https://osf.io/thser/download # download the example data
iss generate --cpus 8 --genomes SRS121011.fasta --model hiseq --output hiseq_reads
If you have created your custom model, give to --model the path of your custom model file:
curl -O -J -L https://osf.io/thser/download # download the example data
iss generate --genomes SRS121011.fasta --model model.npz --output model_reads
If your multi-fasta file contain more genomes than the number of organisms for which you wish to simulate reads, you can use the --n_genomes/-u parameter:
curl -O -J -L https://osf.io/thser/download # download the example data
iss generate --genomes SRS121011.fasta --n_genomes 5 --model novaseq --output novaseq_reads
The above command will pick 5 random genomes in your multi-fasta and generate reads from them.
You can also provide multiple input files:
curl -O -J -L https://osf.io/thser/download # download the example data
curl -O -J -L https://osf.io/37kg8/download # download another example file
iss generate --genomes SRS121011.fasta minigut.fasta --n_genomes 5 --model novaseq --output novaseq_reads
Amplicons¶
To generate amplicon reads, use the --sequence_type amplicon option:
# no example data is provided here
iss generate --genomes my_amplicons.fasta ---readcount_file counts.txt -sequence_type amplicon --model nextseq --output reads
where counts.txt is a tab-delimited file containing the number of reads to generate for each amplicon sequence present in my_amplicons.fasta.
Alternatively, you can use the --n_reads option to generate a fixed number of reads, together with an abundance distribution.
Draft genomes¶
InSilicoseq’s --genomes option assumes complete genomes in multifasta format.
That is, each record in fasta files passed to the --genomes option is treated as a different genome
If you have draft genome files containing contigs, you can give them to the --draft option:
# input file not provided in this example
iss generate --draft my_draft_genome.fasta --model novaseq --output novaseq_reads
Or if you have more than one draft:
# input file not provided in this example
iss generate --draft draft1.fasta draft2.fasta draft3.fasta --model novaseq --output novaseq_reads
You can also combine your drafts with complete genomes:
# input file not provided in this example
iss generate -g complete_genomes.fasta --draft draft.fasta --model novaseq --output novaseq_reads
Required input files¶
By default, InSilicoSeq only requires 1 file in order to start generating reads: 1 (multi-)fasta files containing your input genome(s).
If you don’t want to use a multi-fasta file or don’t have one at hand but are equipped with an Internet connection, you can download random genomes from the ncbi with the --ncbi/-k parameter:
iss generate --ncbi bacteria -u 10 --model miseq --output miseq_ncbi
The above command will generate reads from 10 random bacterial genomes from the NCBI
Additionally, you can supply tab separated kingdoms if you wish to have mixed datasets:
iss generate -k bacteria viruses -u 10 4 --model miseq --output miseq_ncbi
The above command will generate reads from 10 random bacteria and 4 random viruses.
--ncbi/-k accepts the following values: bacteria, viruses and archaea.
In addition the the 2 fastq files and the abundance file, the downloaded genomes will be saved in miseq_ncbi_genomes.fasta in your current directory.
The --ncbi is compatible with --draft and --genomes so you can combine the 3 options.
Abundance distribution¶
With default settings, the abundance of the input genomes is drawn from a log-normal distribution.
Alternatively, you can use other distributions with the --abundance parameter: uniform, halfnormal, exponential or zero-inflated-lognormal
If you wish to fine-tune the distribution of your genomes, InSilicoSeq also accepts an abundance file:
curl -O -J -L https://osf.io/thser/download # download the example data
iss generate -g SRS121011.fasta --abundance_file abundance.txt -m HiSeq -o HiSeq_reads
Example abundance file for a multi-fasta containing 2 genomes: genome_A and genome_B.
genome_A 0.2
genome_B 0.8
For the abundance to make sense, the total abundance in your abundance file must equal 1.
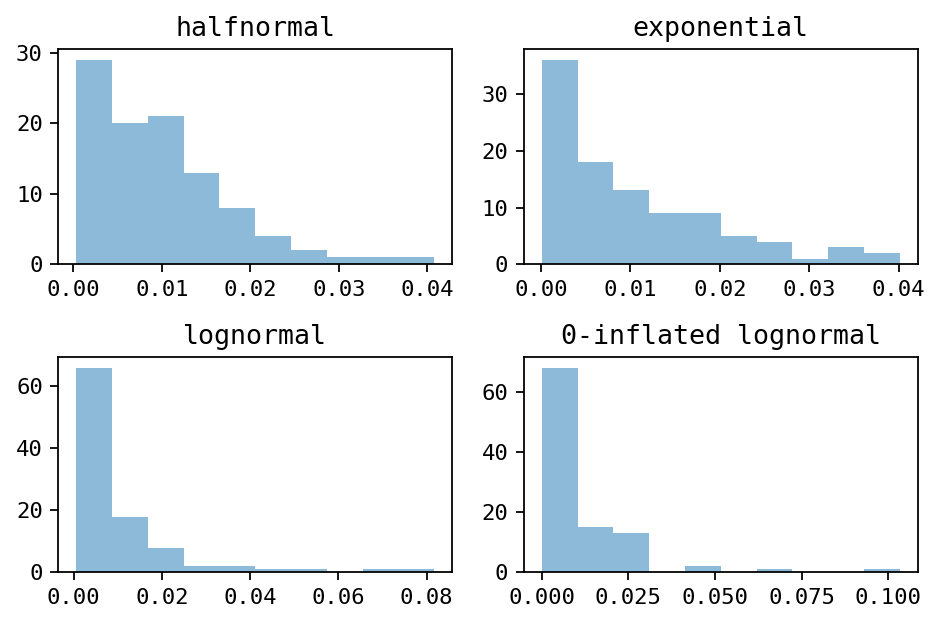
Histograms of the different distribution (drawn with 100 samples)¶
Coverage distribution¶
In the context of InSilicoSeq, the abundance is the proportion of reads in a sample, which since it does not acount for the length of the genome, does not necessarily reflect the number of organisms present in a sample.
The coverage and coverage_file options allow for simulating reads according to a coverage distribution instead of abundance.
iss generate --ncbi bacteria -U 50 --coverage lognormal -n 25M \
--model novaseq --output reads
The coverage_file option works similarly to the abundance_file option.
For two genomes A and B:
iss generate --genomes genomes.fasta --coverage_file coverage.txt \
--model novaseq --output reads
with, for a coverage of 20x for genome_A and 100x for genome_B, the coverage file coverage.txt will be:
genome_A 20
genome_B 100
GC bias¶
InSilicoSeq can also model gc bias:
curl -O -J -L https://osf.io/thser/download # download the example data
iss generate -g SRS121011.fasta --model miseq --gc_bias --output reads
Basic error model¶
By default InSilicoSeq uses Kernel Density Estimators for generating reads. Both the pre-built models (miseq, hiseq and novaseq), as well as the model files you build yourselves are that way.
If you wish to use a much simpler model (because you don’t have the need for insertions and deletion errors per example), you can use --mode basic
curl -O -J -L https://osf.io/thser/download # download the example data
iss generate -g SRS121011.fasta --mode basic --output basic_reads
Full list of options¶
–genomes¶
Input genome(s) from where the reads will originate
–draft¶
Input draft genome(s) from where the reads will originate
–ncbi¶
Download input genomes from RefSeq instead of using –genomes. Requires –n_genomes option. Can be bacteria, viruses, archaea or a combination of the three (space-separated)
–n_genomes¶
How many genomes will be downloaded from the ncbi. Required if –ncbi is set. If more than one kingdom is set with –ncbi, multiple values are necessary (space-separated).
–abundance¶
Abundance distribution (default: lognormal). Can be uniform, halfnormal, exponential, lognormal or zero_inflated_lognormal.
–abundance_file¶
Abundance file for coverage calculations (default: None).
–coverage¶
coverage distribution. Can be uniform, halfnormal, exponential, lognormal or zero-inflated-lognormal.
–coverage_file¶
file containing coverage information (default: None).
–readcount_file¶
file containing read_count information (default: None).
–n_reads¶
Number of reads to generate (default: 1000000). Allows suffixes k, K, m, M, g and G (ex 0.5M for 500000).
–mode¶
Error model. If not specified, using kernel density estimation (default: kde). Can be ‘kde’ or ‘basic’
–model¶
Error model file. (default: None). Use HiSeq, NextSeq, NovaSeq, MiSeq or Miseq-[20,24,28,32] for a pre-computed error model provided with the software, or a file generated with iss model. If you do not wish to use a model, use –mode basic or –mode perfect. The name of the built-in models are case insensitive.
–gc_bias¶
If set, may fail to sequence reads with abnormal GC content. Does not guarantee –n_reads (default: False)
–sequence_type¶
Type of sequencing. Can be metagenomics or amplicon (default: metagenomics).
–cpus¶
Number of cpus to use. (default: 2).
–seed¶
Seed all the random number generators
–quiet¶
Disable info logging
–debug¶
Enable debug logging
–output¶
Output file path and prefix (Required)
–compress¶
Compress the output in gzip format (default: False).
–store_mutations¶
Generates an additional VCF file with the mutations introduced in the reads (bool).